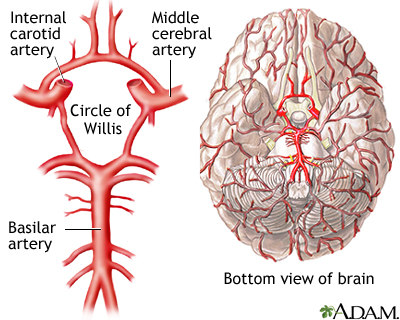
Lá no seu cérebro, ou melhor, aí, no seu cérebro, embaixo dele, existe um complexo de artérias que mais parece o Rodoanel, chamado de polígono de Willis. Forma-se a partir as conexões entre as duas artérias carótidas internas, divididas em artéria cerebral anterior e artéria cerebral média, e a artéria basilar, que através das artérias cerebrais posteriores conectam-se à parte da frente do cérebro. Lá vai uma figura para você não ficar boiando:
 |
| http://www.nlm.nih.gov/medlineplus/ency/images/ency/fullsize/18009.jpg |
Muito bem. Mas quem foi Willis??
Thomas Willis nasceu na Inglaterra em 1621, e morreu em 1675. Era médico e descobridor, em uma época nebulosa da história da medicina, onde pouco se sabia e, por isso, pouco se podia fazer pelas doenças. Graduou-se na famosa e antiga Universidade de Oxford. Foi médico da corte de Charles I da Inglaterra, e rivalizava na Europa, tanto na medicina como na política, com outro Thomas, mas este na Holanda, Thomas Sydenham, que descreveu a coreia de Sydenham da febre reumática.
Entre os vários livros publicados por Thomas Willis, alguns foram sobre neurologia (pelo menos três lidavam com anatomia do cérebro), de tal modo que Thomas Willis, para alguns, é visto como o fundador da neurologia. Ele tinha um vasto conhecimento anatômico, e utilizava-se dele na sua prática clínica.
Thomas Willis foi um pioneiro na descrição anatômica do sistema nervoso, sendo que sua descoberta mais notável foi o famoso Polígono (ou círculo) de Willis, que pode ser visto na figura acima. Em 1664, ele publicou o livro Cerebri anatome, sobre a anatomia do cérebro e dos nervos. É neste trabalho que ele usa pela primeira vez o nome neurologia (neuros + logos, ou estudo do sistema nervoso). Em 1667, ele publicou o livro Pathologicae cerebri, et nervosi generis specimen, um trabalho em patologia e neurofisiologia (ou o pouco que se sabia à época). Seus trabalhos posteriores também foram pioneiros na proclamação da relação entre mente e cérebro, tendo sido, também, um precursor da psiquiatria. Mas apesar de muito escrever, ele chegou a ter condutas estranhas para o tratamento de alguns pacientes, como bater na cabeça dos pacientes com pedaços de madeira, de modo que o que ele escrevia, ele não colocava em prática.
Os nervos cranianos foram primeiramente descritos por Willis, na ordem em que conhecemos hoje (do 1º ao 12º, são Olfatório, Óptico, Oculomotor, Troclear, Trigêmeo, Abducente, Facial, Vestibulococlear, Glossofaríngeo, Vago, Acessório e Hipoglosso) (leia mais aqui).
Ele descreveu o corpo caloso (leia mais aqui) e várias outras estruturas cerebrais, como o fórnix, os corpos mamilares (localizados na base do crânio), o cerebelo, as artérias carótidas e a artéria basilar.
Fora da neurologia, o diabetes mellitus se chama mellitus por causa de Willis (o diabetes era conhecido, antigamente, como doença de Willis), e foi ele que relacionou a urina de gosto doce ao diabetes (não faça isso em casa, mas diabéticos descompensados podem eliminar quantidades de açúcar na urina, o que a deixa com um gosto adocicado).
Thomas Willis:
Thomas Willis:
 |
| http://media-2.web.britannica.com/eb-media/32/40532-004-7E058048.jpg |
