A doença denominada de Neuropatia Periférica Hereditária com Predisposição a Paralisias de Pressão ou HNPP é uma doença geneticamente determinada, tal como a doença de Charcot-Marie-Tooth descrita no post anterior. No entanto, a HNPP é menos frequente e talvez menos conhecida, mas não tão rara. Sua mutação genética encontra-se no mesmo local da mutação da doença de Charcot-Marie-Tooth, mas o tipo da mutação é diferente (a isso chamamos de heterogeneidade fenotípica, ou seja, uma mesma mutação causando vários tipos de problemas diferentes).
A doença foi descrita por DeJong em 1947 em um mineiro de carvão da Holanda que trabalhava agachado, e em 4 de seus parentes de 3 gerações diferentes. Logo, outros pesquisadores publicaram caso parecidos, sempre com história familiar semelhante. Logo em 1965, quando a eletroneuromiografia já existia, as anormalidades de condução nervosa começaram a ser descritas. Os estudos de biopsia logo vieram, mostrando os nervos, após sucessivas perdas de mielina (desmielinização) e novos recapamentos com mielina (remielinização) parecendo bulbos de cebola ou salsichas.
Na figura abaixo você vê nervos cortados de frente. As fibras mais evidentes demonstram bainhas de mielina redundantes e grossas, tendo este aspecto o nome de tomácula, ou bulbo de cebola, que pode ser encontrado nas biopsias de nervo de pacientes com HNPP.
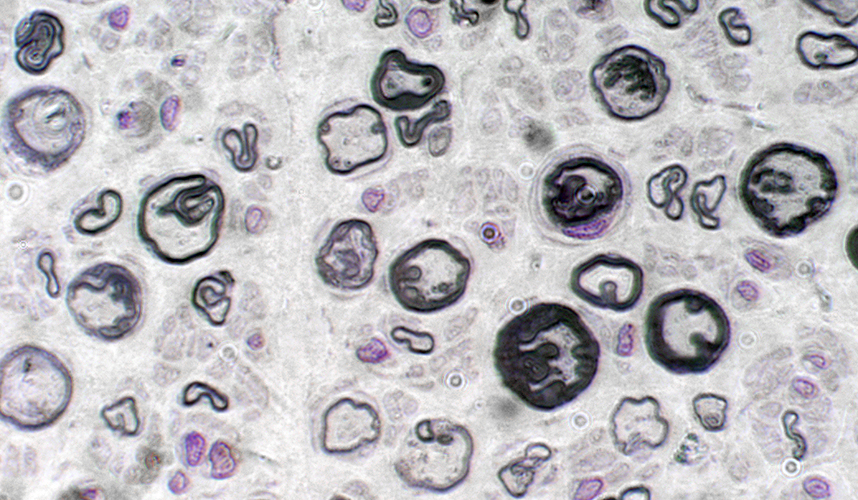 |
| http://neuromuscular.wustl.edu/pics/biopsy/nerve/hnpp/hnpplptb.jpg |
Os estudos genéticos apareceram na década de 80 do século XX, com a categorização dos vários subtipos de doença de Charcot-Marie-Tooth, e posteriormente a categorização genética da HNPP. Descobriu-se que a mutação envolve uma proteína chamada de Proteína da Mielina Periférica, ou PMP22 (22 kiloDáltons é o peso da molécula da proteína). A perda (ou deleção) de uma parte do gene que codifica esta proteína leva à maior parte dos casos de HNPP, mas há outros tipos de mutações do mesmo gene.
A doença geralmente começa aos 20 ou 30 anos de idade, mas pode começar desde a infância até casos onde idosos de 70 anos desenvolveram a doença. A paralisia pode estar presente raramente ao nascimento. A doença, tal como a doença de Charcot-Marie-Tooth, é lentamente progressiva e não começa de forma súbita. Pode ser assintomática em alguns pacientes, ou se, não demonstrar sintomas. Ou pode se tornar aparente (não ser causada, mas ficar mais visível) quando o paciente desenvolve uma neuropatia periférica por outra causa, como diabetes, uso de álcool ou drogas, ou doenças autoimunes.
Em geral, o paciente tem maia sintomas motores que sensitivos. Os pacientes costumam queixar-se de que, ao deixar o membro em repouso, como o braço com o cotovelo em cima da mesa, ou mantendo as pernas cruzadas, acabam por desenvolver paralisias dos nervos lesados e dormências no local de inervação do nervo lesado, que ao invés de durar segundos a minutos como normalmente, duram de semanas a meses. Os ataques de fraqueza e dormência, sempre de nervos específicos, podem ocorrer subitamente, de forma indolor, e com recuperação dos déficits no início da doença.
À medida que os traumas aos nervos vão ocorrendo, déficits neurológicos como perda de força leve ou alterações de sensibilidade podem acabar permanecendo. Muitos destes traumas são mínimos ou inevitáveis, como carrear peso, por exemplo, ou mesmo escrever. Tipicamente, os pacientes não relatam dor. Alguns pacientes podem acabar desenvolvendo uma polineuropatia, ou seja, um acometimento de vários nervos, geralmente à medida que a doença avança e os traumas se sucedem.
Em um estudo de 1996, 40% dos pacientes não sabiam que tinham a doença mesmo apresentando sintomas, e 25% eram assintomáticos. Em outro estudo com 70 paciente de 1999, o número de ataques que os pacientes apresentavam em toda a vida, em média, era de 2. Os nervos mais afetados, do mais para o menos afetado, são o nervo fibular, o ulnar, o plexo braquial, o nervo radial e o mediano (veja abaixo):
 |
| http://files.fisiogrup.webnode.com.br/200000068-cf17ad10b5/nervo_ulnar_all.jpg |
Na figura acima, vemos o nervo ulnar, o nervo mediano e o plexo braquial, identificado como Nerve Roots (Raízes Nervosas).
 |
| http://www.sistemanervoso.com/images/anatomia/np_i_102.jpg |
Esta bela figura aí em cima mostra o nervo fibular, na parte de fora da perna (você pode palpá-lo, com cuidado, tocando o ossinho que fica do lado de fora do joelho e logo abaixo dele; o nervo está atrás deste osso).
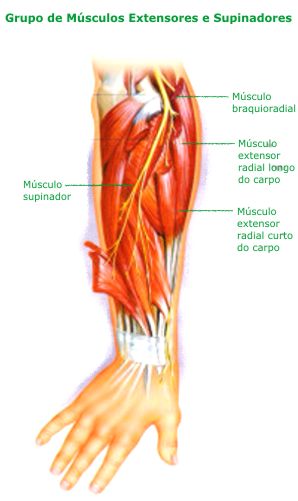 |
| http://www.clinicadevita.com.br/imagens/supinador_01.jpg |
E este aí em cima é o nervo radial, cuja paralisia dá a famosa mão caída.
O mesmo estudo citado acima demonstra que as paralisas duraram, em média, mais de 3 meses em 15% dos pacientes. Alguns poucos pacientes podem ter uma forma semelhante à doença de Charcot-Marie-Tooth (leia o post anterior para conhecer esta doença).
Alguns pacientes sem sintomas pode apresentar anormalidades ao exame neurológico ou na eletroneuromiografia. A doença é rara na infância. Dois autores relataram em 2003 um caso de uma criança de 2 anos com a doença.
A doença pode, no entanto, ser complicada por outras doenças que cursam com neuropatia periférica, como diabetes, problemas de rim (uremia), uso de álcool e outras drogas, deficiências de vitamina, e uso de medicações que causam neuropatia periférica. Nestes pacientes, a doença pode apresentar-se de forma mais grave, pode complicar as outras causas de neuropatias periféricas, e pode evoluir de forma mais agressiva.
O diagnóstico deve sempre se basear em suspeita diagnóstica pelo médico, já que esta é uma doença incomum entre as causas de neuropatias periféricas. No entanto, a presença de familiares afetados chama a atenção. Mas o médico deve, também, afastar causas tratáveis de neuropatias periféricas, já que a HNPP é genética.
O uso da eletroneuromiografia deve sempre ser considerado no diagnóstico de neuropatia periférica. Já testes genéticos e biopsia muscular devem ter suas indicações discutidas com os médicos que assistem o paciente.
Não há tratamento específico para a HNPP, por ser uma doença genética (ainda não, mas terapias gênicas estão em estudo). No entanto, os pacientes devem procurar evitar situações onde há chance de haver paralisias de nervos, como cruzar as pernas, por exemplo. Os pacientes devem evitar ou tratar as doenças que cursam com neuropatias periféricas, como o diabetes, a deficiência e vitaminas, o uso de álcool e o uso de medicações que pioram neuropatias periféricas (discutidas no post anterior). O uso de medicações e vitaminas deve ser feito somente com prescrição médica.
A doença pode, no entanto, ser complicada por outras doenças que cursam com neuropatia periférica, como diabetes, problemas de rim (uremia), uso de álcool e outras drogas, deficiências de vitamina, e uso de medicações que causam neuropatia periférica. Nestes pacientes, a doença pode apresentar-se de forma mais grave, pode complicar as outras causas de neuropatias periféricas, e pode evoluir de forma mais agressiva.
O diagnóstico deve sempre se basear em suspeita diagnóstica pelo médico, já que esta é uma doença incomum entre as causas de neuropatias periféricas. No entanto, a presença de familiares afetados chama a atenção. Mas o médico deve, também, afastar causas tratáveis de neuropatias periféricas, já que a HNPP é genética.
O uso da eletroneuromiografia deve sempre ser considerado no diagnóstico de neuropatia periférica. Já testes genéticos e biopsia muscular devem ter suas indicações discutidas com os médicos que assistem o paciente.
Não há tratamento específico para a HNPP, por ser uma doença genética (ainda não, mas terapias gênicas estão em estudo). No entanto, os pacientes devem procurar evitar situações onde há chance de haver paralisias de nervos, como cruzar as pernas, por exemplo. Os pacientes devem evitar ou tratar as doenças que cursam com neuropatias periféricas, como o diabetes, a deficiência e vitaminas, o uso de álcool e o uso de medicações que pioram neuropatias periféricas (discutidas no post anterior). O uso de medicações e vitaminas deve ser feito somente com prescrição médica.
Nenhum comentário:
Postar um comentário
Comente na minha página do Facebook - Dr Flávio Sekeff Sallem,
Médico Neurologista