Post adaptado do site Medlink Neurology.
No nosso segundo post sobre distúrbios psiquiátricos, vamos falar das desordens que fazem parte do espectro autista.
No nosso segundo post sobre distúrbios psiquiátricos, vamos falar das desordens que fazem parte do espectro autista.
Entre 1908 e 1911, o psiquiatra suíço Eugen Bleuler usou o termo "autístico" para descrever uma forma de doença descrita por ele, a esquizofrenia.
O termo Autismo provem do grego "autos", ou seja, "a si mesmo". Em 1943, Leo Kanner, psiquiatra americano, descreveu termos relacionados ao autismo, como a solidão, a dificuldade de relacionamento com outros e os déficits de comunicação. Em 1944, o famoso pediatra austríaco Hans Asperger descreveu 4 crianças com dificuldades de interação social.
A definição atual de autismo e desordens do espectro autista passou por modificações desde a primeira edição do Diagnostic and Statistical Manual of Mental Disorders, o DSM, a Bíblia da psiquiatria, de 1952. De lá para cá, já somam-se 6 edições do DSM, a última sendo a 5ª edição, de 2003 (Há a 1ª, a 2ª, a 3ª, a 4ª, a 4ªrevisada (TR) e a 5ª). O DSM-IV-TR de 2000 traz o nome "Desordens Pervasivas do Desenvolvimento" (Pervasivo significa difuso), englobando o Autismo, a Síndrome de Asperger, Desordens de Desenvolvimento Não Especificadas, a Desordem Desintegrativa da Infância e a Síndrome de Rett.
O DSM-V engloba todas estas definições na Desordem do Espectro Autista, que passaremos a chamar de DEA.
DEA é uma desordem de desenvolvimento neurocognitivo caracterizada por prejuízo da comunicação e interação sociais, interesses isolados, comportamentos repetitivos, e hipo ou hiperatividade a estímulos sensoriais. Estes e outros sintomas podem começar a aparecer os 6 aos 18 meses de idade. Indicadores precoces incluem ausência de expressões de alegria, ausência de gesticulação, ausência de compartilhamento da alegria, ausência de resposta ao ser chamado pelo nome, e movimentos repetitivos do corpo, braços, mãos ou dedos.
Outras características observadas em crianças mais velhas são:
1. Falta de contato visual ou expressão facial
2. Falta de interesse em estar com outras crianças, prefere brincar só
3. Não se relaciona com amigos ou semelhantes da mesma idade
4. Não compartilha alegria e excitação ou conquistas com os outros
5. Indiferente às tristezas e sentimentos dos outros
6. Dificuldade de ver o ponto de vista do outro
7. Trata outros como objetos ou ferramentas (p.ex., usar a mão da mãe para virar a maçaneta)
8. Coloca os brinquedos em fila ou presta atenção somente em uma parte do brinquedo
9. Não consegue imaginar o faz-de-conta
10. Interesse diminuído por jogos em grupo
11. Comportamentos estereotipados, como balançar as mãos, balançar o corpo ou girar em um mesmo lugar
12. Interesse extremo em assuntos isolados, como pontes ou trens
13. Estressa-se facilmente com pequenas modificações na rotina
14. Hipo ou hipersensibilidade a estímulos sensitivos, como ruídos
Os indivíduos com DEA podem até afeiçoar-se a alguém ou desejar a interação social, mas falta-lhes as habilidades necessárias para isso. As crianças podem ter um desenvolvimento típico ou prejuízos significantes da linguagem, tanto para expressar-se como para entender. Os pacientes podem não se engajar em uma comunicação significativa, e podem não oferecer resposta verbal, o que leva a um prejuízo em manter um diálogo. A linguagem falada por ser estereotipada (ou seja, o paciente pode usar sempre as mesma palavras ou fraes) e pode apresentar ecolalia (ou seja, ele repete o que os outros disseram).
Cerca de 33% das crianças com DEA têm algum tipo de prejuízo intelectual (retardo mental). Além disso, pacientes com DEA podem apresentar hiperatividade, impulsividade, falta de atenção (não confundir com TDAH, discutido no post anterior). Pode haver problemas de humor, afeto, e ansiedade, com medo excessivo ou medo de mudança. As resposta a ameaças podem ser inapropriadas (ou não reagem ou reagem de maneira excessiva). Surtos de agressividade ou de comportamento auto-mutilante (a criança lesa a si mesma), como morder-se ou bater a cbaeça na parede, podem ocorrer.
Cerca de 1/4 das crianças com autismo têm epilepsia, o que é muito maior que a frequência de epilepsia na população geral, com a maioria das crises começando após os 10 anos de idade. Além disso, essas crianças podem apresntar mais frequentemente constipação intestinal e problemas com alimentação (dificuldades em se alimentar ou comer somente alimentos escolhidos por elas). Dor abdominal, excesso de gases, diarreia e náuseas podem ocorrer com frequência, podendo estes sintomas estar associados a ansiedade excessiva. Por último, crianças com DEA podem ter insônia ou sono entrecortado (acordar várias vezes à noite), e aparentemente déficits de sono nestes pacientes associam-se a baixo escore de inteligência, dificuldades com linguagem verbal (uso de palavras para se expressar), dificuldades com habilidades do dia-a-dia, déficit de sociabilidade e outros problemas.
A DEA é uma das doenças do desenvolvimento mais comuns em crianças, sendo que nos EUA, este complexo acomete 1 em cada 88 crianças, sendo mais comum em meninos que em meninas. A doença parece, pelos estudos, não ter preferência por classe social ou etnia.
Não há uma causa única para o DEA, e através de estudos genéticos, cerca de 15 a 20% dos casos poderão ter uma causa documentada. Na maioria dos casos, a causa permanece desconhecida. Várias doenças neurológicas e não neurológicas podem levar a DEA, como a esclerose tuberosa (uma doença do sistema nervoso em que há a formação de tumores cerebrais e lesões típicas de pele, podendo estar associada a retardo mental e crises epilépticas), a síndrome do X frágil e anormalias dos cromossomos, além de várias outras doenças.
Há concordância de 60% entre gêmeos monozigóticos e 0% entre gêmeos dizigóticos (ou seja, gêmeos idênticos compartilham a DEA em 60% dos casos, e gêmeos não idênticos compartilham raramente a doença), indicando a forte interação genética na determinação da doença. Mas provavelmente muitos são os genes envolvidos, o que torna o diagnóstico ainda mais difícil. Fora isso, filhos de pais com DEA têm 90% de chance de desenvolver a doença.
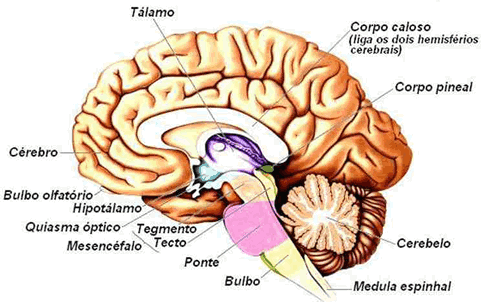
Da mesma forma que com o TDAH (leia aqui), há anormalidades cerebrais que parecem se correlacionar com os casos de autismo e doenças correlatas. Estudos sugerem envolvimento dos lobos da parte da frente do cérebro (os lobos frontais) além da amígdala e do cerebelo.
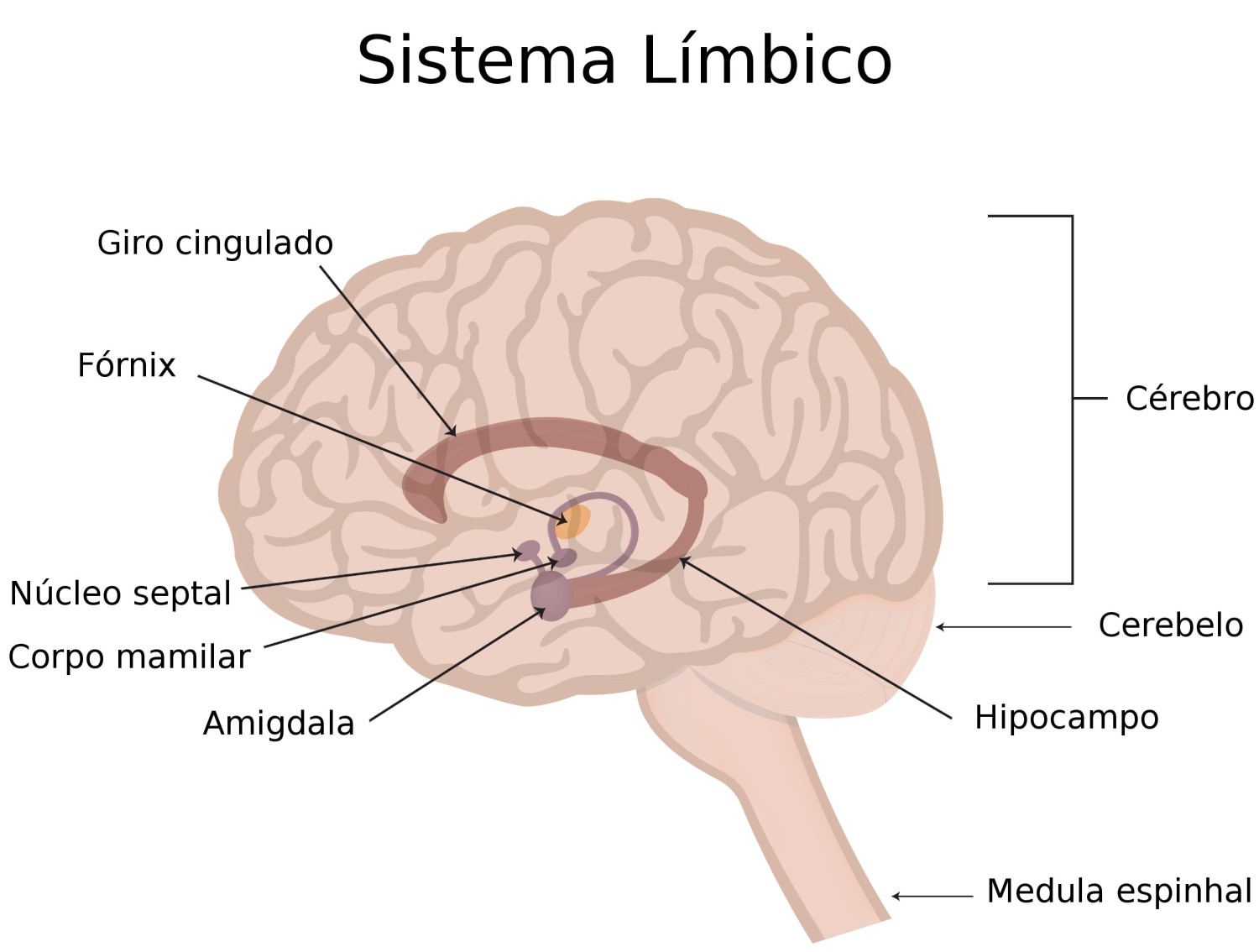
Bem, seu cérebro tem uma amígdala (não somente sua garganta, como um dos meus irmãos, há vários anos, pensou quando soube que havia uma doença cerebral que afetava a amígdala... cerebral!). A amígdala funciona juntamente com o sistema límbico, o sistema responsável, entre outras coisas, pelas suas emoções. Veja abaixo:
A amígdala é a parte final de um núcleo cerebral muito importante, o núcleo caudato. Mas voltemos à DEA.
Há sugestão de alterações do córtex cerebral (aumento do volume de substância cinzenta) em várias regiões cerebrais como o lobo frontal, e diminuição do volume das partes mais posteriores do cérebro, os lobos occipital e temporal. É interessante que 1/3 dos autismas tem aumento do tamanho e do volume cerebral.
Bem, falamos bastante coisa sobre o que é o autismo e as desordens relacionadas, seus sintomas, sua epidemiologia básica e as alterações mais simples encontradas nos cérebros de pacientes com essas desordens. E o diagnóstico? Como é feito?
Inicialmente, há a necessidade de afastarmos outras doenças, especialmente as que podem ser tratadas com mais facilidade. E para isso, há a necessidade de um neuropediatra ou um psiquiatra que conheça a doença, seus sintomas e os diagnósticos diferenciais, ou seja, as doenças que se parecem, mas não são. Entre os diagnósticos diferenciais de DEA, temos:
1. Surdez infantil
2. Desordens de desenvolvimento da linguagem
3. Retardo mental
4. Esquizofrenia
5. Transtorno obssessivo-compulsivo
6. Síndrome de tiques motores complexos (falaremos sobre isso em post posterior, mas há uma breve discussão sobre isso no post sobre TDAH).
Outros diagnósticos são complexos demais para serem discutidos aqui, especialmente por um neurologista.
E há exames para detectar DEA no meu filho(a)? Bem, vamos iniciar essa discussão da seguinte maneira:
O primeiro passo para um diagnóstico é a história da doença, desde os primeiros meses de vida. História de infecções da mãe durante a gestação, lesões que possam, porventuram, ter acontecido com a criança durante ou após o parto, meningite, encefalite na criança. Saber se há história familiar de autismo, doenças genéticas, desordens de desenvolvimento ou doenças psiquiátricas na família, especialmente em irmãos, é importante, pois como já discutido antes, pode haver hereditariedade.
O exame físico da criança passa por saber se há sinais neurológicos que indiquem uma doença, ou características físicas, especialmente da face, que falem a favor de alguma doença genética. Um exame mental da criança, checando interesses, relacionamento social e familiar, comunicação verbal e não verbal, imaginação, comportamentos estranhos ou anormais, dificuldades atencionais, agressividade, auto-mutilação. Há questionários apropriados para isso em consultórios especializados, orientados por médicos.
Claro que outros exames podem ser feitos, mas para descartar outras doenças, como uma avaliação autidiva completa, testes genéticos quando indicados, um eletroencefalograma se houver história de crises epilépticas, pesquisa de aminoácidos e carboidratos na urina atrás de doenças do metabolismo. Ressonância magnética ou tomografia de crânio só devem ser solicitadas em casos específicos, quando há suspeita de lesão cerebral, exame neurológicos com sinais anormais, ou com indicação médica.
Alguns pacientes com DEA pode ter vida social e profissional ativas. Há, inclusive, uma mulher de 67 anos, americana, portadora de autismo chamada Mary Temple Gradin, que mesmo com suas limitações, criadora de várias invenções na área da pecuária, especialmente um meio "humanizado" de abeter gado para carne. Com mais de 400 artigos publicados na área, Temple Gradin, inclusive, teve um filme sobre sua vida, lançado em 2010 (leia mais sobre ela aqui).
Há evidências que sugerem que intervenções intensas, tanto na área médica como na área cognitiva, com reabilitação, estímulos e tratamento médico, começando a partir dos 3 anos de idade (ou seja, com diagnóstico precoce) são um fator chave na promoção de resultados positivos em crianças com autismo, o que torna o prognóstico dessas crianças melhor que pensado antes.
Estas intervenções quais são devem ser discutidas e comandadas pelo pediatra/neuropediatra/psiquiatra infantil que acompanha o paciente.
O apoio familiar é muito importante no cuidado dessas crianças. Os pais e irmãos precisam conhecer a doença e o comportamento da criança, e muitas vezes há a necessidade de apoio profissional nisso.
Por último, há medicações que podem ser usadas para melhorar a atenção e estimular a criança, ao mesmo tempo controlando seus impulsos e comportamentos agressivos. Mas essas medicações devem ser discutidas em consultório, com o especialista.
1. Falta de contato visual ou expressão facial
2. Falta de interesse em estar com outras crianças, prefere brincar só
3. Não se relaciona com amigos ou semelhantes da mesma idade
4. Não compartilha alegria e excitação ou conquistas com os outros
5. Indiferente às tristezas e sentimentos dos outros
6. Dificuldade de ver o ponto de vista do outro
7. Trata outros como objetos ou ferramentas (p.ex., usar a mão da mãe para virar a maçaneta)
8. Coloca os brinquedos em fila ou presta atenção somente em uma parte do brinquedo
9. Não consegue imaginar o faz-de-conta
10. Interesse diminuído por jogos em grupo
11. Comportamentos estereotipados, como balançar as mãos, balançar o corpo ou girar em um mesmo lugar
12. Interesse extremo em assuntos isolados, como pontes ou trens
13. Estressa-se facilmente com pequenas modificações na rotina
14. Hipo ou hipersensibilidade a estímulos sensitivos, como ruídos
Os indivíduos com DEA podem até afeiçoar-se a alguém ou desejar a interação social, mas falta-lhes as habilidades necessárias para isso. As crianças podem ter um desenvolvimento típico ou prejuízos significantes da linguagem, tanto para expressar-se como para entender. Os pacientes podem não se engajar em uma comunicação significativa, e podem não oferecer resposta verbal, o que leva a um prejuízo em manter um diálogo. A linguagem falada por ser estereotipada (ou seja, o paciente pode usar sempre as mesma palavras ou fraes) e pode apresentar ecolalia (ou seja, ele repete o que os outros disseram).
Cerca de 33% das crianças com DEA têm algum tipo de prejuízo intelectual (retardo mental). Além disso, pacientes com DEA podem apresentar hiperatividade, impulsividade, falta de atenção (não confundir com TDAH, discutido no post anterior). Pode haver problemas de humor, afeto, e ansiedade, com medo excessivo ou medo de mudança. As resposta a ameaças podem ser inapropriadas (ou não reagem ou reagem de maneira excessiva). Surtos de agressividade ou de comportamento auto-mutilante (a criança lesa a si mesma), como morder-se ou bater a cbaeça na parede, podem ocorrer.
Cerca de 1/4 das crianças com autismo têm epilepsia, o que é muito maior que a frequência de epilepsia na população geral, com a maioria das crises começando após os 10 anos de idade. Além disso, essas crianças podem apresntar mais frequentemente constipação intestinal e problemas com alimentação (dificuldades em se alimentar ou comer somente alimentos escolhidos por elas). Dor abdominal, excesso de gases, diarreia e náuseas podem ocorrer com frequência, podendo estes sintomas estar associados a ansiedade excessiva. Por último, crianças com DEA podem ter insônia ou sono entrecortado (acordar várias vezes à noite), e aparentemente déficits de sono nestes pacientes associam-se a baixo escore de inteligência, dificuldades com linguagem verbal (uso de palavras para se expressar), dificuldades com habilidades do dia-a-dia, déficit de sociabilidade e outros problemas.
A DEA é uma das doenças do desenvolvimento mais comuns em crianças, sendo que nos EUA, este complexo acomete 1 em cada 88 crianças, sendo mais comum em meninos que em meninas. A doença parece, pelos estudos, não ter preferência por classe social ou etnia.
Não há uma causa única para o DEA, e através de estudos genéticos, cerca de 15 a 20% dos casos poderão ter uma causa documentada. Na maioria dos casos, a causa permanece desconhecida. Várias doenças neurológicas e não neurológicas podem levar a DEA, como a esclerose tuberosa (uma doença do sistema nervoso em que há a formação de tumores cerebrais e lesões típicas de pele, podendo estar associada a retardo mental e crises epilépticas), a síndrome do X frágil e anormalias dos cromossomos, além de várias outras doenças.
Há concordância de 60% entre gêmeos monozigóticos e 0% entre gêmeos dizigóticos (ou seja, gêmeos idênticos compartilham a DEA em 60% dos casos, e gêmeos não idênticos compartilham raramente a doença), indicando a forte interação genética na determinação da doença. Mas provavelmente muitos são os genes envolvidos, o que torna o diagnóstico ainda mais difícil. Fora isso, filhos de pais com DEA têm 90% de chance de desenvolver a doença.
Da mesma forma que com o TDAH (leia aqui), há anormalidades cerebrais que parecem se correlacionar com os casos de autismo e doenças correlatas. Estudos sugerem envolvimento dos lobos da parte da frente do cérebro (os lobos frontais) além da amígdala e do cerebelo.
Bem, seu cérebro tem uma amígdala (não somente sua garganta, como um dos meus irmãos, há vários anos, pensou quando soube que havia uma doença cerebral que afetava a amígdala... cerebral!). A amígdala funciona juntamente com o sistema límbico, o sistema responsável, entre outras coisas, pelas suas emoções. Veja abaixo:
 |
| https://psych-brain-trust.wikispaces.com/file/view/amygdala_pic.jpg/261622100/amygdala_pic.jpg |
Há sugestão de alterações do córtex cerebral (aumento do volume de substância cinzenta) em várias regiões cerebrais como o lobo frontal, e diminuição do volume das partes mais posteriores do cérebro, os lobos occipital e temporal. É interessante que 1/3 dos autismas tem aumento do tamanho e do volume cerebral.
Bem, falamos bastante coisa sobre o que é o autismo e as desordens relacionadas, seus sintomas, sua epidemiologia básica e as alterações mais simples encontradas nos cérebros de pacientes com essas desordens. E o diagnóstico? Como é feito?
Inicialmente, há a necessidade de afastarmos outras doenças, especialmente as que podem ser tratadas com mais facilidade. E para isso, há a necessidade de um neuropediatra ou um psiquiatra que conheça a doença, seus sintomas e os diagnósticos diferenciais, ou seja, as doenças que se parecem, mas não são. Entre os diagnósticos diferenciais de DEA, temos:
1. Surdez infantil
2. Desordens de desenvolvimento da linguagem
3. Retardo mental
4. Esquizofrenia
5. Transtorno obssessivo-compulsivo
6. Síndrome de tiques motores complexos (falaremos sobre isso em post posterior, mas há uma breve discussão sobre isso no post sobre TDAH).
Outros diagnósticos são complexos demais para serem discutidos aqui, especialmente por um neurologista.
E há exames para detectar DEA no meu filho(a)? Bem, vamos iniciar essa discussão da seguinte maneira:
O primeiro passo para um diagnóstico é a história da doença, desde os primeiros meses de vida. História de infecções da mãe durante a gestação, lesões que possam, porventuram, ter acontecido com a criança durante ou após o parto, meningite, encefalite na criança. Saber se há história familiar de autismo, doenças genéticas, desordens de desenvolvimento ou doenças psiquiátricas na família, especialmente em irmãos, é importante, pois como já discutido antes, pode haver hereditariedade.
O exame físico da criança passa por saber se há sinais neurológicos que indiquem uma doença, ou características físicas, especialmente da face, que falem a favor de alguma doença genética. Um exame mental da criança, checando interesses, relacionamento social e familiar, comunicação verbal e não verbal, imaginação, comportamentos estranhos ou anormais, dificuldades atencionais, agressividade, auto-mutilação. Há questionários apropriados para isso em consultórios especializados, orientados por médicos.
Claro que outros exames podem ser feitos, mas para descartar outras doenças, como uma avaliação autidiva completa, testes genéticos quando indicados, um eletroencefalograma se houver história de crises epilépticas, pesquisa de aminoácidos e carboidratos na urina atrás de doenças do metabolismo. Ressonância magnética ou tomografia de crânio só devem ser solicitadas em casos específicos, quando há suspeita de lesão cerebral, exame neurológicos com sinais anormais, ou com indicação médica.
Alguns pacientes com DEA pode ter vida social e profissional ativas. Há, inclusive, uma mulher de 67 anos, americana, portadora de autismo chamada Mary Temple Gradin, que mesmo com suas limitações, criadora de várias invenções na área da pecuária, especialmente um meio "humanizado" de abeter gado para carne. Com mais de 400 artigos publicados na área, Temple Gradin, inclusive, teve um filme sobre sua vida, lançado em 2010 (leia mais sobre ela aqui).
Há evidências que sugerem que intervenções intensas, tanto na área médica como na área cognitiva, com reabilitação, estímulos e tratamento médico, começando a partir dos 3 anos de idade (ou seja, com diagnóstico precoce) são um fator chave na promoção de resultados positivos em crianças com autismo, o que torna o prognóstico dessas crianças melhor que pensado antes.
Estas intervenções quais são devem ser discutidas e comandadas pelo pediatra/neuropediatra/psiquiatra infantil que acompanha o paciente.
O apoio familiar é muito importante no cuidado dessas crianças. Os pais e irmãos precisam conhecer a doença e o comportamento da criança, e muitas vezes há a necessidade de apoio profissional nisso.
Por último, há medicações que podem ser usadas para melhorar a atenção e estimular a criança, ao mesmo tempo controlando seus impulsos e comportamentos agressivos. Mas essas medicações devem ser discutidas em consultório, com o especialista.